A fibrose cística em crianças é uma doença genética (hereditária) crônica, progressiva e frequentemente fatal. A fibrose cística afeta principalmente os sistemas respiratório e digestivo. As glândulas sudoríparas e o sistema reprodutivo também são afetados.
Causas genéticas
A fibrose cística em crianças é causada por um gene defeituoso que controla a absorção de sal pelo corpo. Durante a doença, muito sal e pouca água entram nas células do corpo.
 Isso transforma os fluidos que normalmente "lubrificam" nossos órgãos em muco espesso e pegajoso. Esse muco bloqueia as vias respiratórias nos pulmões e obstrui o lúmen das glândulas digestivas.
Isso transforma os fluidos que normalmente "lubrificam" nossos órgãos em muco espesso e pegajoso. Esse muco bloqueia as vias respiratórias nos pulmões e obstrui o lúmen das glândulas digestivas.
O principal fator de risco para o desenvolvimento de fibrose cística é a história familiar, especialmente se um dos pais for portador. O gene que causa a fibrose cística é recessivo.
Isso significa que, para ter essa doença, as crianças devem herdar duas cópias do gene, uma da mãe e do pai. Quando uma criança herda apenas uma cópia, ela não desenvolve fibrose cística. Mas esse bebê ainda será um portador e poderá passar o gene para sua prole.
Os pais portadores do gene da fibrose cística costumam ser saudáveis e assintomáticos, mas passarão o gene para seus filhos.
Na verdade, de acordo com várias estimativas, até 10 milhões de pessoas podem ser portadoras do gene da fibrose cística e não saber sobre ele. Se a mãe e o pai tiverem um gene defeituoso para a fibrose cística, então eles terão uma probabilidade de 1: 4 de ter um filho com fibrose cística.
Sintomas
Os sinais de fibrose cística são múltiplos e podem mudar com o tempo. Normalmente, os sintomas em crianças aparecem pela primeira vez em uma idade muito precoce, mas às vezes eles aparecem um pouco mais tarde.
Embora esta doença cause vários problemas de saúde graves, afeta principalmente os pulmões e o sistema digestivo. Portanto, a forma pulmonar e intestinal da doença é alocada.
Métodos diagnósticos modernos podem detectar fibrose cística em recém-nascidos usando testes de triagem especiais antes que qualquer sintoma apareça.
- 15 - 20% dos recém-nascidos com fibrose cística apresentam obstrução meconial ao nascimento. Isso significa que o intestino delgado está obstruído com mecônio, as fezes originais. Normalmente, o mecônio sai sem problemas. Mas em bebês com fibrose cística, é tão denso e denso que o intestino simplesmente não consegue removê-lo. Como resultado, as alças intestinais se torcem ou não se desenvolvem adequadamente. O mecônio também pode bloquear o cólon e, nessa situação, o bebê não evacuará por um ou dois dias após o nascimento.
- Os próprios pais podem notar alguns dos sinais de fibrose cística em recém-nascidos. Por exemplo, quando a mãe e o pai beijam um bebê, eles percebem que sua pele tem gosto de sal.
- A criança não está ganhando peso corporal suficiente.
- A icterícia pode ser outro sinal precoce de fibrose cística, mas esse sintoma não é confiável, pois muitos bebês desenvolvem essa condição imediatamente após o nascimento e geralmente se resolve após alguns dias por conta própria ou com a ajuda de fototerapia. É mais provável que a icterícia, neste caso, esteja associada a fatores genéticos, e não à fibrose cística. O rastreamento permite que os médicos façam um diagnóstico preciso.
- O muco pegajoso produzido por esta doença pode causar sérios danos aos pulmões. Crianças com fibrose cística freqüentemente desenvolvem infecções no peito porque esse fluido espesso forma um terreno fértil para o crescimento de bactérias. Qualquer criança com essa condição sofre de uma série de tosses graves e infecções brônquicas. Sibilância intensa e falta de ar são problemas adicionais de que os bebês sofrem.
Embora esses problemas de saúde não sejam exclusivos de crianças com fibrose cística e possam ser tratados com antibióticos, as consequências em longo prazo são graves. Afinal, a fibrose cística pode causar tantos danos aos pulmões de uma criança que eles não podem funcionar adequadamente.
- Algumas crianças com fibrose cística desenvolvem pólipos nas passagens nasais. Os bebês podem ter sinusite aguda ou crônica grave.
- O sistema digestivo é outra área onde a fibrose cística se torna a principal causa de danos. Assim como o muco pegajoso bloqueia os pulmões, ele também causa problemas semelhantes em diferentes partes do sistema gastrointestinal. Isso interfere na passagem suave dos alimentos pelos intestinos e na capacidade do sistema de digerir os nutrientes. Como resultado, os pais podem perceber que seu filho não está ganhando peso ou crescendo normalmente. As fezes do bebê cheiram mal e parecem brilhantes devido à má digestão de gorduras. Crianças (geralmente com mais de quatro anos) às vezes sofrem de intussuscepção. Quando isso acontece, uma parte do intestino é inserida em outra. O intestino se dobra telescopicamente em si mesmo, como uma antena de televisão.
- O pâncreas também é afetado. Freqüentemente, ocorre inflamação nele. Essa condição é conhecida como pancreatite.
- Tosse freqüente ou fezes difíceis às vezes causam prolapso retal. Isso significa que uma parte do reto se projeta ou sai do ânus. Cerca de 20% das crianças com fibrose cística apresentam essa condição. Em alguns casos, o prolapso retal é o primeiro sinal perceptível de fibrose cística.
Assim, se uma criança tem fibrose cística, ela pode ter as seguintes manifestações e sintomas, que podem ser leves e graves:
 tosse ou respiração ofegante;
tosse ou respiração ofegante;- a presença de grande quantidade de muco nos pulmões;
- infecções pulmonares freqüentes, como pneumonia e bronquite;
- falta de ar;
- pele salgada;
- crescimento atrofiado, mesmo com bom apetite;
- obstrução de mecônio em recém-nascidos;
- Fezes frequentes que são soltas, grandes ou gordurosas
- dor de estômago ou inchaço.
 tosse ou respiração ofegante;
tosse ou respiração ofegante;Diagnóstico
Quando os sintomas começam a aparecer, a fibrose cística não é, na maioria dos casos, o primeiro diagnóstico do médico. Existem muitos sintomas de fibrose cística e nem todas as crianças apresentam todos os sintomas.
Outro fator é que a doença pode variar de leve a grave em diferentes crianças. A idade em que os sintomas aparecem também muda. Alguns foram diagnosticados com fibrose cística na infância, enquanto outros foram diagnosticados mais tarde na vida. Se a doença for leve, a criança pode não ter problemas até a adolescência ou mesmo a idade adulta.
Exame genético
Ao passar nos testes genéticos durante a gravidez, os pais agora podem descobrir se seus futuros filhos podem ter fibrose cística. Mas mesmo quando os testes genéticos confirmam a presença de fibrose cística, ainda não há como prever com antecedência se os sintomas de uma criança em particular serão graves ou leves.

O teste genético também pode ser feito após o nascimento do bebê. Uma vez que a fibrose cística é uma condição hereditária, o médico pode sugerir testes nos irmãos do bebê, mesmo se eles não apresentarem sintomas. Outros membros da família, especialmente primos, também devem ser testados.
Geralmente, o bebê é testado para fibrose cística se nascer com íleo meconial.
Amostra de suor
Após o nascimento, o teste diagnóstico padrão para fibrose cística é o teste do suor. É um método de diagnóstico preciso, seguro e indolor. O estudo usa uma pequena corrente elétrica para estimular as glândulas sudoríparas com pilocarpina. Isso estimula a produção de suor. Dentro de 30-60 minutos, o suor é coletado em papel de filtro ou gaze e verificado os níveis de cloreto.
Uma criança deve fazer um teste de cloreto no suor de mais de 60 em duas amostras de suor separadas para ser diagnosticada com fibrose cística. Os valores normais de suor para bebês são mais baixos.
Determinação de tripsinogênio
O teste pode não ser informativo em recém-nascidos, pois eles não produzem suor suficiente. Nesse caso, outro tipo de teste pode ser utilizado, como a determinação do tripsinogênio imunorreativo. Nesse teste, o sangue coletado 2 a 3 dias após o nascimento é analisado para uma proteína específica chamada tripsinogênio. Os resultados positivos devem ser confirmados pelo teste do suor e outros testes. Além disso, uma pequena porcentagem de crianças com fibrose cística apresenta níveis normais de cloreto no suor. Eles só podem ser diagnosticados por testes químicos para a presença do gene mutado.
Outros tipos de exame
Alguns dos outros testes que podem ajudar a diagnosticar a fibrose cística são radiografias de tórax, testes de função pulmonar e análise de escarro. Eles mostram como os pulmões, o pâncreas e o fígado estão funcionando. Isso ajuda a determinar a extensão e a gravidade da fibrose cística, uma vez que é diagnosticada.
Esses testes incluem:
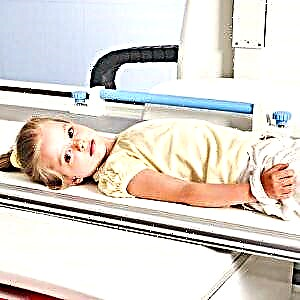 Raio-x do tórax;
Raio-x do tórax;- exames de sangue para avaliar a quantidade de nutrientes que entram no corpo;
- estudos bacterianos que confirmam o crescimento de Pseudomonas aeruginosa e Staphylococcus aureus ou outras bactérias hemofílicas nos pulmões. Essas bactérias são comuns na fibrose cística, mas podem não afetar pessoas saudáveis;
- testes de função pulmonar para medir o efeito da fibrose cística na respiração.
Os testes de função pulmonar são realizados quando a criança tem idade suficiente para colaborar nos testes;
- análise de fezes. Isso ajudará a identificar anormalidades gastrointestinais características da fibrose cística.
 Raio-x do tórax;
Raio-x do tórax;Tratamento da fibrose cística em crianças
- Visto que a fibrose cística é uma doença genética, a única maneira de prevenir ou curar é usar a engenharia genética desde cedo. Idealmente, a terapia gênica pode reparar ou substituir um gene defeituoso. Neste estágio do desenvolvimento da ciência, esse método permanece irreal.
- Outra opção de tratamento é dar à criança com fibrose cística uma forma ativa de um produto proteico que é insuficiente ou não está no corpo. Infelizmente, isso também não é viável.
Portanto, no momento, nem a terapia gênica nem qualquer outro tratamento radical para a fibrose cística são conhecidos pela medicina, embora as abordagens baseadas em drogas estejam agora sendo estudadas.
Nesse ínterim, o melhor que os médicos podem fazer é amenizar os sintomas da fibrose cística ou retardar a progressão da doença para melhorar a qualidade de vida da criança. Isso é realizado com terapia antibiótica combinada com procedimentos para remover o muco espesso dos pulmões.
A terapia é adaptada às necessidades de cada criança. Para crianças com doença muito progressiva, o transplante de pulmão pode ser uma alternativa.
Anteriormente, a fibrose cística era uma doença fatal. As melhores terapias desenvolvidas nos últimos 20 anos aumentaram o tempo médio de vida das pessoas com fibrose cística para 30 anos.
Tratamento de doenças pulmonares
A área de tratamento mais importante para a fibrose cística é a luta contra a falta de ar, que causa infecções pulmonares frequentes. Fisioterapia, exercícios e medicamentos são usados para aliviar bloqueios da mucosa nas vias aéreas do pulmão.
 drenagem brônquica (postural). É realizada colocando o paciente em uma posição que permita a drenagem do muco dos pulmões, ou seja, o nível dos ombros fica abaixo do nível da região lombar. Durante o procedimento, o tórax ou as costas são batidos e vibrados para ativar o muco e ajudá-lo a sair das vias aéreas. Esse processo se repete em diferentes áreas do tórax para reduzir a quantidade de muco em diferentes áreas de cada pulmão. Este procedimento deve ser realizado todos os dias. E é muito importante ensinar essa técnica aos familiares;
drenagem brônquica (postural). É realizada colocando o paciente em uma posição que permita a drenagem do muco dos pulmões, ou seja, o nível dos ombros fica abaixo do nível da região lombar. Durante o procedimento, o tórax ou as costas são batidos e vibrados para ativar o muco e ajudá-lo a sair das vias aéreas. Esse processo se repete em diferentes áreas do tórax para reduzir a quantidade de muco em diferentes áreas de cada pulmão. Este procedimento deve ser realizado todos os dias. E é muito importante ensinar essa técnica aos familiares;- Os exercícios ajudam a limpar o muco e estimular a tosse para limpar os pulmões. Também pode melhorar a condição física geral da criança;
- de medicamentos, os medicamentos em aerossol são usados para facilitar a respiração.
 drenagem brônquica (postural). É realizada colocando o paciente em uma posição que permita a drenagem do muco dos pulmões, ou seja, o nível dos ombros fica abaixo do nível da região lombar. Durante o procedimento, o tórax ou as costas são batidos e vibrados para ativar o muco e ajudá-lo a sair das vias aéreas. Esse processo se repete em diferentes áreas do tórax para reduzir a quantidade de muco em diferentes áreas de cada pulmão. Este procedimento deve ser realizado todos os dias. E é muito importante ensinar essa técnica aos familiares;
drenagem brônquica (postural). É realizada colocando o paciente em uma posição que permita a drenagem do muco dos pulmões, ou seja, o nível dos ombros fica abaixo do nível da região lombar. Durante o procedimento, o tórax ou as costas são batidos e vibrados para ativar o muco e ajudá-lo a sair das vias aéreas. Esse processo se repete em diferentes áreas do tórax para reduzir a quantidade de muco em diferentes áreas de cada pulmão. Este procedimento deve ser realizado todos os dias. E é muito importante ensinar essa técnica aos familiares;Esses medicamentos incluem:
- broncodilatadores que alargam as vias aéreas;
- mucolíticos que afinam o muco;
- descongestionantes que reduzem o inchaço nas vias aéreas
- antibióticos para combater infecções pulmonares. Eles podem ser administrados por via oral, na forma de aerossol ou injetados na veia.
Tratamento de problemas digestivos
Os problemas digestivos na fibrose cística são menos graves e mais fáceis de controlar do que os problemas pulmonares.
Uma dieta balanceada com alto teor calórico é freqüentemente prescrita com baixo teor de gordura e rica em proteínas e enzimas pancreáticas que auxiliam na digestão.
Os suplementos de vitamina A, D, E e K são indicados para uma boa nutrição. Enemas e mucolíticos são usados para tratar a obstrução intestinal.
Cuidando de uma criança com fibrose cística
Quando você é informado de que seu bebê tem fibrose cística, você precisa tomar medidas extras para garantir que o recém-nascido receba os nutrientes de que necessita e que as vias respiratórias permaneçam desobstruídas e saudáveis.
Alimentando
Para ajudar na digestão adequada, você precisará dar ao seu bebê os suplementos de enzimas prescritos pelo médico no início de cada mamada.
Visto que crianças pequenas comem com frequência, você deve sempre levar enzimas e comida para bebê com você.

Os sinais de que seu filho pode precisar de enzimas ou de um ajuste de dose de enzima incluem:
- incapacidade de ganhar peso apesar de um forte apetite;
- fezes frequentes, gordurosas e malcheirosas;
- inchaço ou gás.
Crianças com fibrose cística precisam de mais calorias do que outras crianças de sua faixa etária. A quantidade de calorias extras de que precisam varia de acordo com a função pulmonar de cada criança, o nível de atividade física e a gravidade da doença.
As necessidades calóricas de uma criança podem ser ainda maiores durante a doença. Mesmo uma infecção leve pode aumentar drasticamente a ingestão de calorias.
A fibrose cística também perturba a função normal das células que constituem as glândulas sudoríparas da pele. Como resultado, os bebês perdem muito sal quando suam, levando a um alto risco de desidratação. Qualquer ingestão adicional de sal deve ser administrada conforme recomendado por um especialista.
Educação e desenvolvimento
Pode-se esperar que a criança se desenvolva de acordo com a norma. Quando uma criança entra no jardim de infância ou na escola, ela pode receber um Plano de Educação Individualizado de acordo com a Lei de Educação de Pessoas com Deficiências.
Um plano individualizado garante que seu filho possa continuar seus estudos se adoecer ou for para o hospital, e também inclui as providências necessárias para visitar uma instituição educacional (por exemplo, fornecer tempo adicional para lanches).
Muitas crianças com fibrose cística continuam a aproveitar a infância e a crescer levando uma vida plena. À medida que a criança cresce, ela pode precisar de muitos procedimentos médicos e, ocasionalmente, vai ao hospital.
A criança deve ser encorajada a ser o mais ativa possível. Seu filho pode precisar de ajuda extra de um pai para se ajustar à escola e à vida cotidiana.A transição da infância para a idade adulta também pode ser desafiadora, pois a criança deve aprender a tratar a fibrose cística por conta própria.
Acima de tudo, as crianças com fibrose cística e suas famílias devem manter uma atitude positiva. Os cientistas continuam a fazer avanços significativos na compreensão das anormalidades genéticas e fisiológicas na fibrose cística e no desenvolvimento de novas abordagens de tratamento, como a terapia genética. Existe a perspectiva de melhorar ainda mais o atendimento aos pacientes com fibrose cística e até mesmo de descobrir um tratamento!